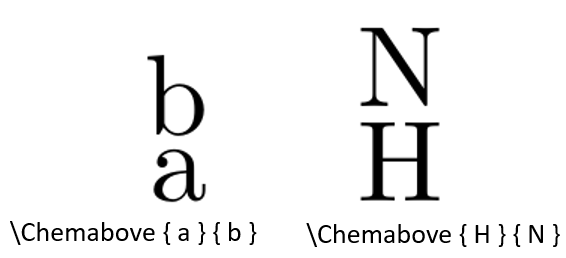Dataset Description
The proposed handwritten dataset is named EDU-CHEMC and it consists of 60,974 handwritten molecular structure images collected in educational scenarios. These images were obtained using various devices such as cameras, scanners, and screens. The key characteristics of this dataset are as follows:
Real-world Educational Scenarios
The dataset includes handwritten molecular structures from primary and secondary education scenarios, which, to our knowledge, have never been made publicly available before. Additionally, a small proportion of the data comprises people copying and photographing ChEMBL molecular structures, also under real settings.
Combination of Molecular Structures and Formulas
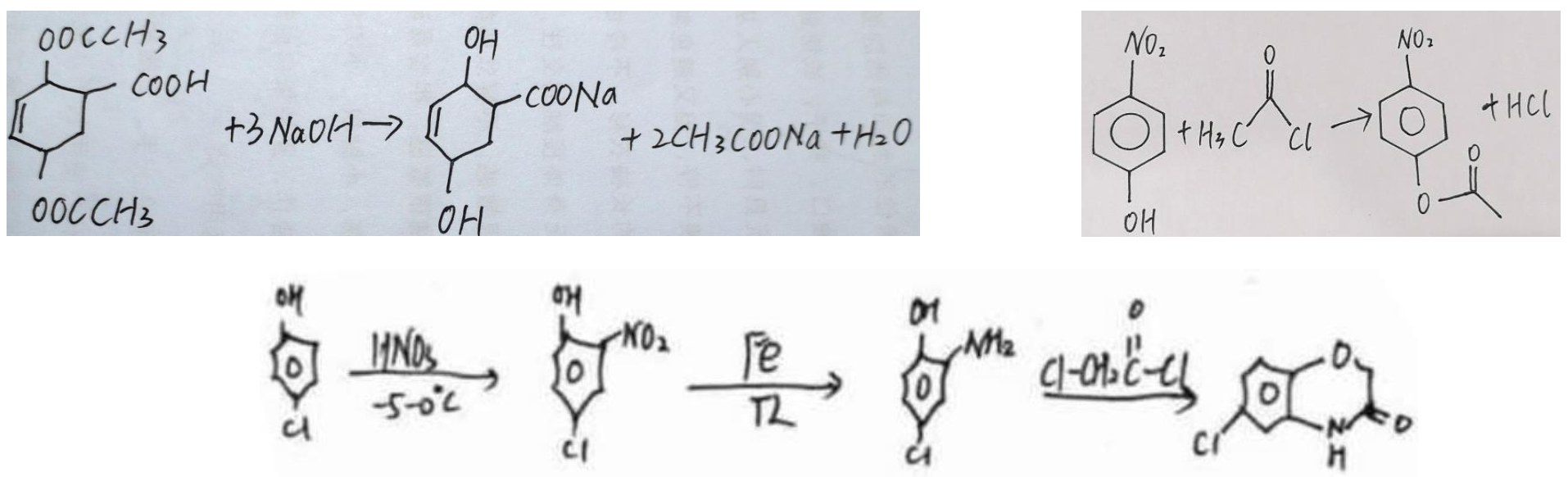
In addition to isolated molecular structures, the dataset also includes instances where formulas are combined with molecular structures. This is demonstrated by organic reaction equations, as shown in Figure 1.
Diversity in Writing Styles
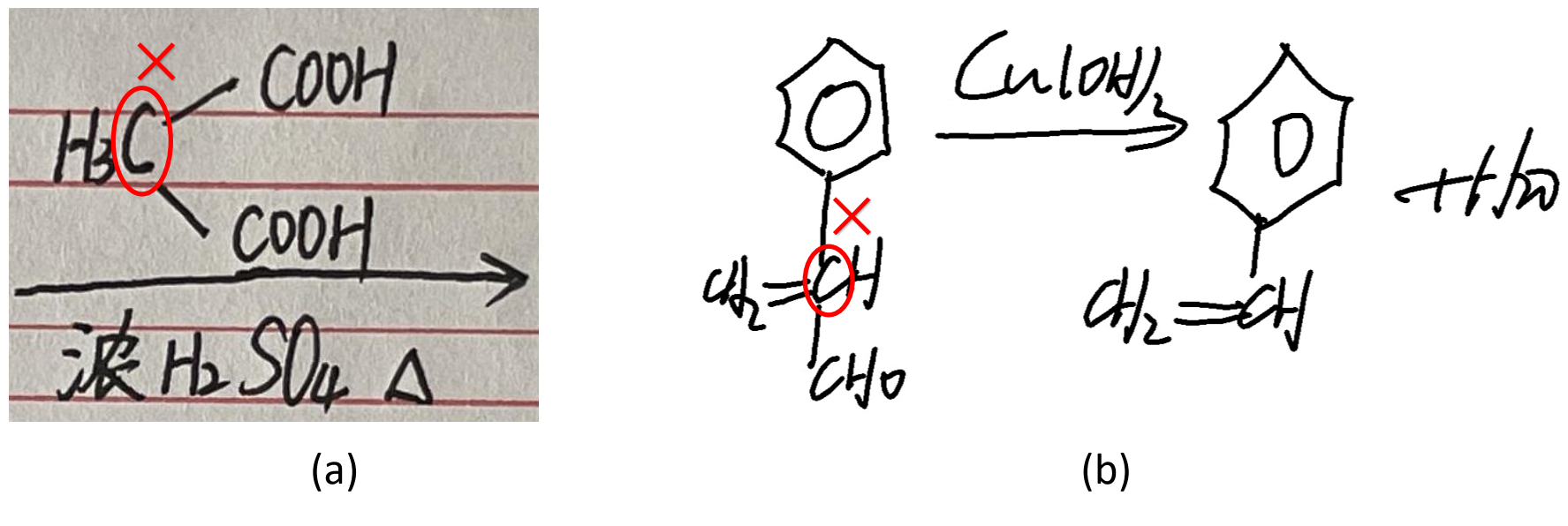
The dataset exhibits a variety of handwriting styles, encompassing variations in the utilization of abbreviations, Kekulé ring notations for benzene, as well as the presence or absence of hydrogen atoms. Notably, it also comprises instances of erroneous or nonexistent structures that violate chemical principles, as demonstrated in Figure 2, thereby emphasizing its potential application in educational correction and revision.
Complex Molecular Structures
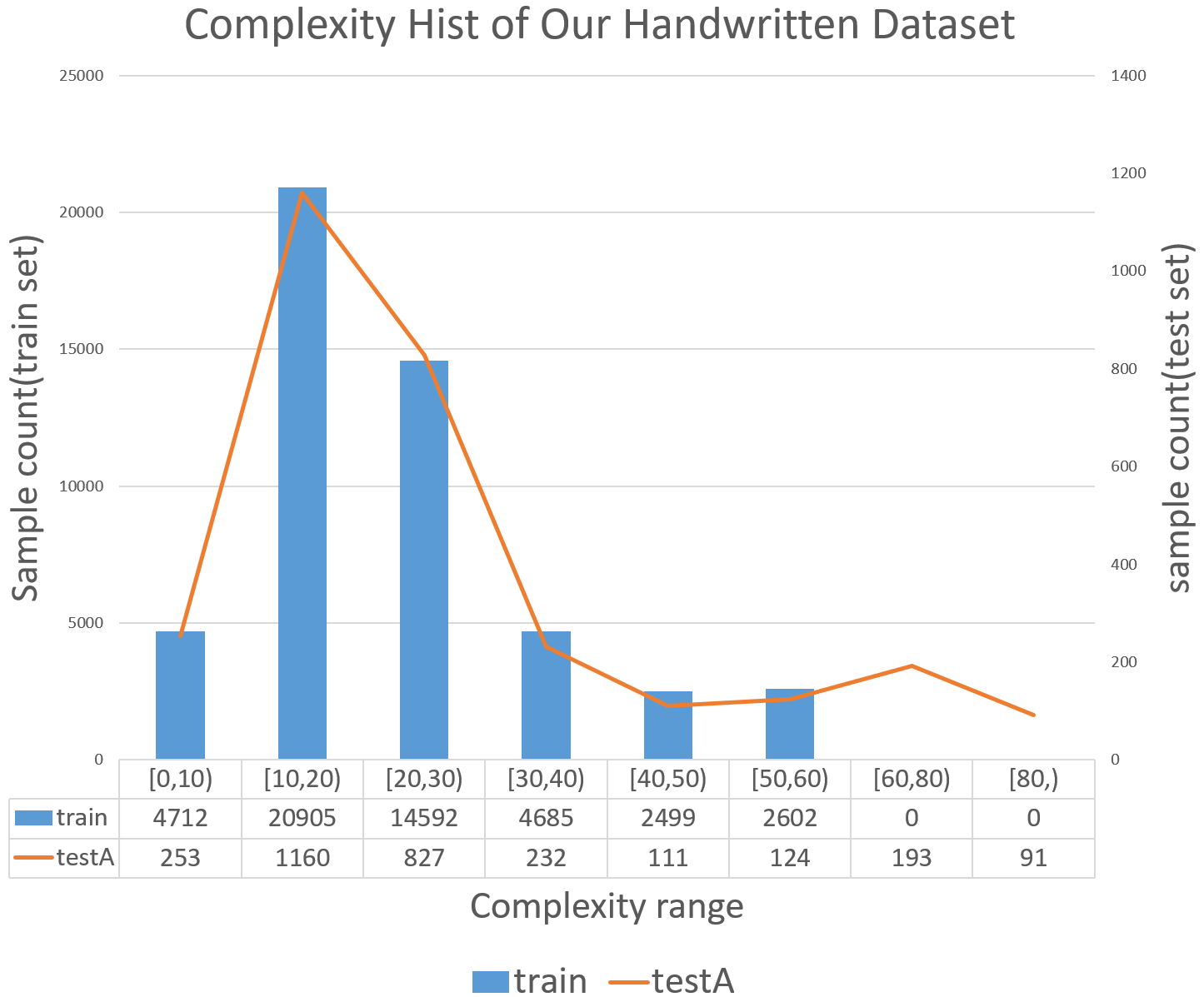
The complexity of molecular structures in the dataset is quantified by the total number of atoms and bonds. Approximately 10% of the testing set comprises structures with a complexity level exceeding the most intricate examples in the training set, as illustrated in Figure 3.
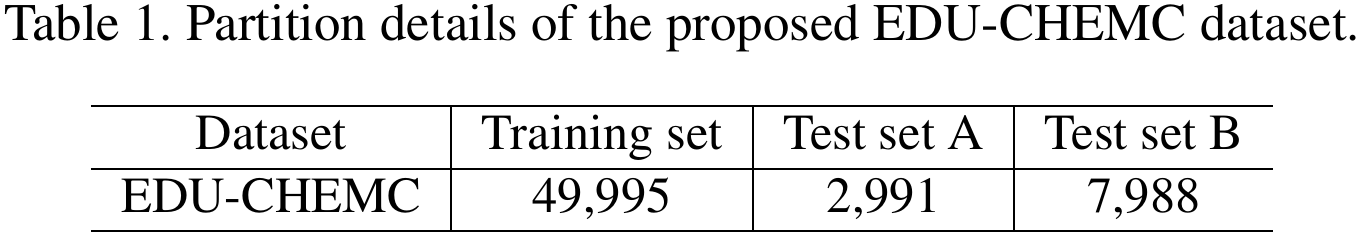
The data partitions of the EDU-CHEMC dataset are outlined as follows:
Annotation
The training set includes an annotation json file for each image, sharing the same filename. Each file consists of three components:
- chemfig
- ssml_normed
- ssml_sd
A raw, human-annotated Chemfig string that can be rendered with texlive using the chemfig package. Note that this string is purely for reference.
A normed version of the Chemfig string, removing complex syntax and retaining only basic rules for ease of program parsing and readability. Participants must submit results in this format, and it is the only one supported by the Parsing Tool and Graph Matching Tool.
The training target of our baseline. Compared to "ssml_normed", bond unit angles are quantified with step of 15 degree, while bond lengths are omitted as most are equal to 1. It's directly applicable to sequence-to-sequence model training. For additional information, refer to this paper.
Guidance for SSML-Normed
Here are some guidelines for SSML-Normed.
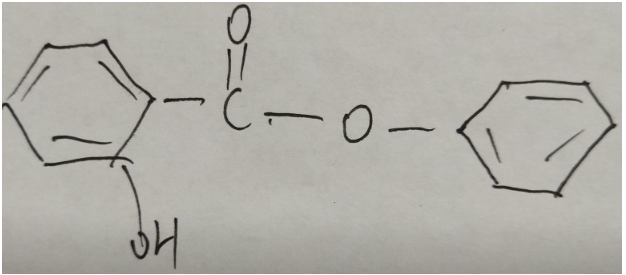
The ssml-normed string comprises four primary units: "Atom", "‘Bond", "Branch", and "Reconnection". Consider a chemical structure as a graph, where atoms signify nodes and bonds represent edges. Two atoms can connect via a bond, thus a chemical graph is essentially made up of atoms and bonds.
To derive an ssml-normed string from a chemical graph, follow these steps:
- Traverse the entire graph using depth-first search (DFS) and record the atoms and bonds in the order they were traversed.
- Convert each atom or bond into string tokens. The rules will be discussed later.
- Include branch tokens during traversing to represent multiple branches from an atom.
- Incorporate reconnection marks to indicate reconnection bonds. The concept of reconnection will be clarified later.
- After completing the traversal, concatenate all tokens to form the ssml-normed string.
Atom
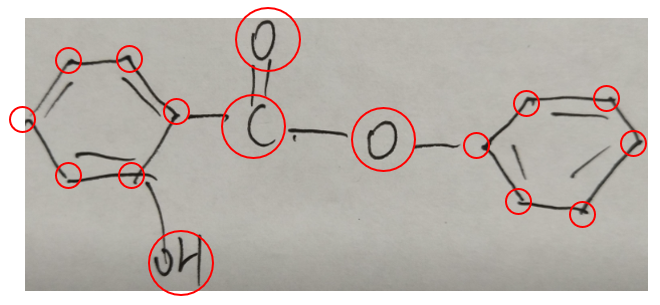
Atoms appear at two endpoints of each bond. In an image, an atom's content may include several characters (like "OH", "C" in Figure 4) or no characters (like the atom on a benzene ring). Figure 5 gives an example of all atoms in a chemical structure image.
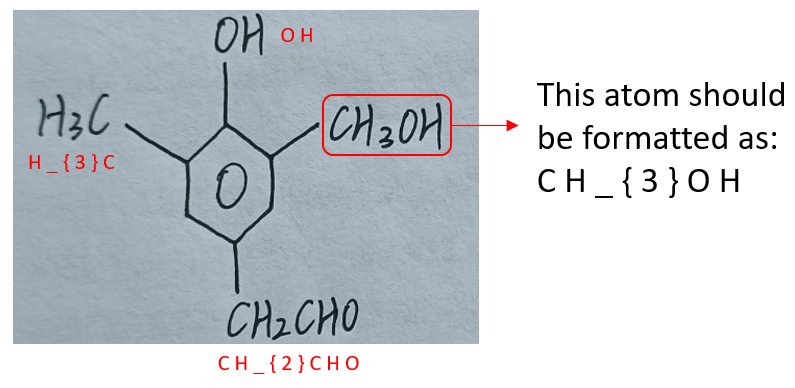
To format an atom to string tokens, simply append all its characters to the output sequence. If the atom's content includes formulas, format it according to latex rules, such as "C H _ { 2 }". See Figure 6 for more examples of atom formatted results.
Bond
Bonds are chemical bonds in an image. They should be formatted according to chemfig syntax rules in one of the following ways:
<bond_type>[:<angle>,<length>]
<bond_type>[:<angle>]
Note that a bond is always formatted into one string token.
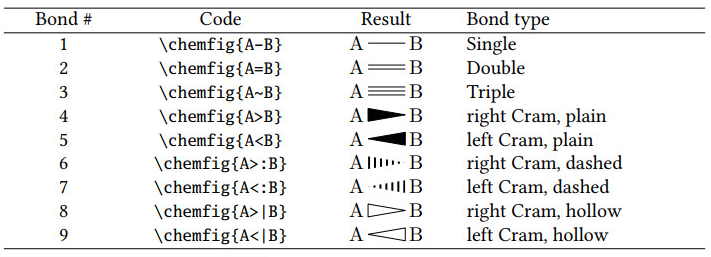
<bond_type>: Nine types are depicted in Figure 7.
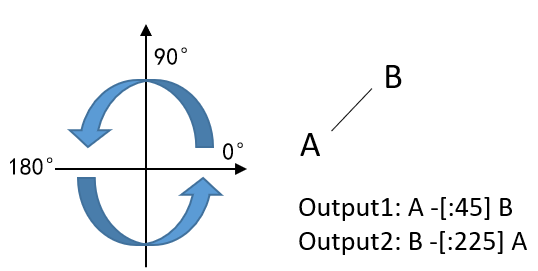
<angle>: When two atoms A and B are connected by a bond C, and formatted in the order "A", "C", "B", the angle of bond C refers to the angle of a directed line segment from point A to B in a polar coordinate system. Angles increase counterclockwise and are relative to the horizontal.
<length>: This is the relative bond length. The default value is 1. Generally, all bonds in a chemical structure have the same length so we can set all bond’s relative length to 1. But if some bonds are noticeably shorter or longer, you can set their length to a value larger or smaller than 1 to maintain the correct structure.
Branch
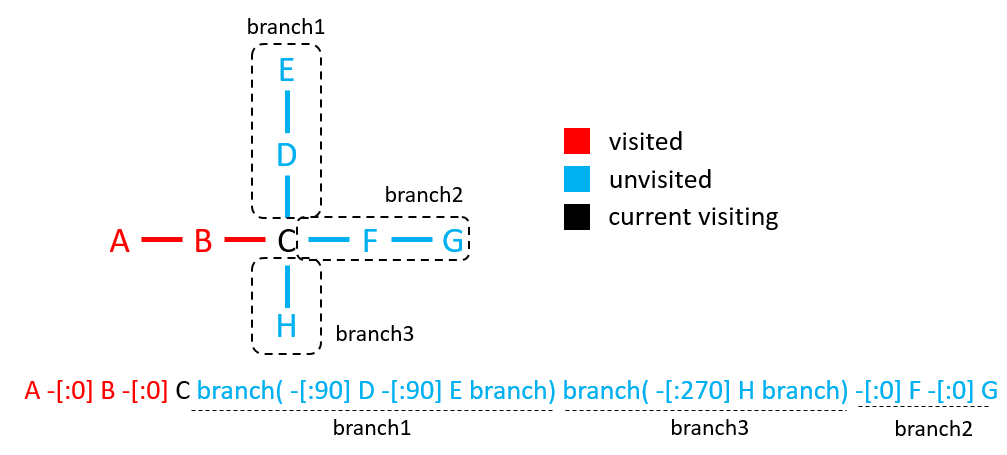
Branches occur during a DFS traversal of a graph when there's more than one unvisited bond attached to an atom, which can be chosen as next element to continue traversal. Each candidate bond represents a branch. Once a candidate bond is chosen,begin a branch with the special token "branch(" and format the chosen bond. Tokens following it are within the branch until no more bonds can be chosen. The branch ends with the token "branch)". If a visiting atom has N branches, only the content of the first N-1 branches being explored should be placed within a pair of branch tokens. It is optional whether the content of last branch being explored should be placed within branch tokens.
Figure 9 illustrates branch syntax rules. Here, Atom "C" has 3 branches explored in the order of branch1, branch3, and branch2. The ssml-normed string is displayed at the bottom.
Reconnection
Reconnection occurs during traversal when a currently visiting atom A is connected to a previously visited atom B at more than two timesteps before by a bond C. We define that there is a reconnection between atom A and atom B. Since atom B can not be visited twice, we use reconnection marks to represent the connection relationship between atom A and B instead of appending "A", "C", and "B" to the output sequence.
Reconnection marks are:
Reconnection start: ?[<name>]
Reconnection end: ?[<name>,{<bond_type>}]
<name> is a short string of English letters, while <bond_type> indicates the bond connecting the atoms (see Figure 7). Reconnection marks are attached to atoms. A reconnection mark is placed after the tokens of a certain atom in the output sequence. Two atoms with same <name> are considered to reconnect with each other through a bond of type <bond_type>.
When reconnecting atom A and B, designate one as the start and the other as the end. The end marks must contain <bond_type> while the start cannot. An atom can be attached with multiple reconnection end marks but at most one reconnection start marks. It is recommended to choose the first visited one to be the reconnection start.
For directed bond types such as "<", ">", the reconnection bond's correct direction is from reconnection start atom to reconnection end atom. For example, "B ?[a]" and "A ?[a, {>}]" equals to "B >[:
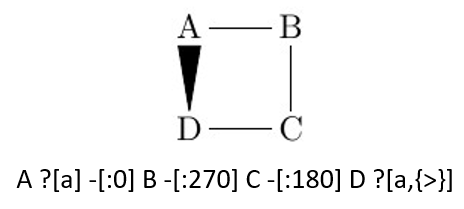
Special Tokens
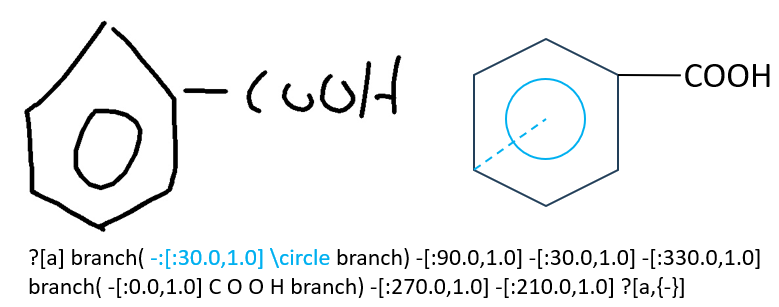
\circle
We use "\circle" to denote the special atom (circle) in benzene. To indicate the connection between the circle and an arbitrary atom on the ring, we utilize a virtual bond with the type "-:". During submission, you only need to randomly select one atom on the ring to connect with the circle atom, as our graph matching tool will normalize this case.
\Chemabove
"\Chemabove { a } { b }" means character "b" is positioned above the character "a". This syntax is commonly used to format some special atoms such as "NH", Refer to Figure 12 for an example.